|
Anaphase-promoting complex Anaphase-promoting complex (also called the cyclosome or APC/C) is an E3 ubiquitin ligase that marks target cell cycle proteins for degradation by the 26S proteasome. The APC/C is a large complex of 11–13 subunit proteins, including a cullin (Apc2) and RING (Apc11) subunit much like SCF. Other parts of the APC/C have unknown functions but are highly conserved.[1] It was the discovery of the APC/C (and SCF) and their key role in eukaryotic cell-cycle regulation that established the importance of ubiquitin-mediated proteolysis in cell biology. Once perceived as a system exclusively involved in removing damaged protein from the cell, ubiquitination and subsequent protein degradation by the proteasome is now perceived as a universal regulatory mechanism for signal transduction whose importance approaches that of protein phosphorylation. In 2014, the APC/C was mapped in 3D at a resolution of less than a nanometre, which also uncovered its secondary structure. This finding could improve understanding of cancer and reveal new binding sites for future cancer drugs.[2][3] Function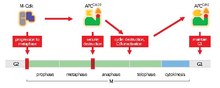 The APC/C's main function is to trigger the transition from metaphase to anaphase by tagging specific proteins for degradation. The three major targets for degradation by the APC/C are securin and S and M cyclins. Securin releases separase, a protease, when degraded. Separase then triggers the cleavage of cohesin, the protein complex that binds sister chromatids together. During metaphase, sister chromatids are linked by intact cohesin complexes. When securin undergoes ubiquitination by the APC/C and releases separase, which degrades cohesin, sister chromatids become free to move to opposite poles for anaphase. The APC/C also targets the mitotic cyclins for degradation, resulting in the inactivation of M-CDK (mitotic cyclin-dependent kinase) complexes, promoting exit from mitosis and cytokinesis.[1] Unlike the SCF, activator subunits control the APC/C. Cdc20 and Cdh1 are the two activators of particular importance to the cell cycle. These proteins target the APC/C to specific sets of substrates at different times in the cell cycle, thus driving it forward. The APC/C also plays an integral role in the maintenance of chromatin metabolism, particularly in G1 and G0, and plays a key role in phosphorylation of H3 through destruction of the aurora A kinase.[4] The critical substrates of the APC/C appear to be securin and the B type cyclins. This is conserved between mammals and yeast. In fact, yeast are viable in the absence of the APC/C if the requirement for targeting these two substrates is eliminated.[5] SubunitsThere is not a vast amount of extensive investigation on APC/C subunits, which serve mostly as adaptors. Studies of APC subunits are mainly conducted in yeast, and studies show that the majority of yeast APC subunits are also present in vertebrates, this suggests conservation across eukaryotes. Eleven core APC subunits have been found in vertebrates versus thirteen in yeast.[1] Activator subunits bind to APC at varying stages of the cell cycle to control its ubiquitination activity, often by directing APC to target substrates destined for ubiquitination. The specificity of APC ligases is proposed to be controlled by the incorporation of specificity factors into the ligase complex, instead of substrate phosphorylation. i.e.: The subunit, CDC20 allows APC to degrade substrates such as anaphase inhibitors (Pdsp1) at the beginning of anaphase, on the other hand when CDC20 is substituted for specificity factor Hct1, APC degrades a different set of substrates, particularly mitosis cyclins in late anaphase. Activators CDC20 and Cdh1 are of particular significance and are the most widely studied and familiar of the APC/C subunits. The catalytic core of the APC/C consists of the cullin subunit Apc2 and RING H2 domain subunit Apc11. These two subunits catalyze ubiquitination of substrates when the C-terminal domain of Apc2 forms a tight complex with Apc11. RING/APc11 binds to the E2-ubiquitin conjugate that catalyzes the transfer of ubiquitin to an active site in E2.[1] In addition to the catalytic functionality, other core proteins of the APC are composed multiple repeat motifs with the main purpose of providing molecular scaffold support. These include Apc1, the largest subunit which contains 11 tandem repeats of 35–40 amino acid sequences, and Apc2, which contains three cullin repeats of approximately 130 amino acids total.[6] The major motifs in APC subunits include tetratricopeptide (TPR) motifs and WD40 repeats 1.[1] C-termini regions of CDC20 and Cdh1 have a WD40 domain that is suggested to form a binding platform that binds APC substrates, thus contributing to APCs ability to target these substrates, although the exact mechanism through which they increase APC activity is unknown.[7] It is also suggested that variations in these WD40 domains result in varying substrate specificity, which is confirmed by recent results suggesting that different APC substrates can directly and specifically bind to Cdc20 and Cdh1/Hct1 Ultimately, the specificity differences are responsible for the timing of the destruction of several APC targets during mitosis. With CDC20 targeting a few major substrates at metaphase and Cdh1 targeting a broader range of substrates towards late mitosis and G1.[8] Most notably, 4 subunits of yeast APC/C consist almost entirely of multiple repeats of the 34 amino acid tetratricopeptide residue (TPR) motif. These TPR subunits, Cdc16,[9] Cdc27,[10] Cdc23, and Apc5, mainly provide scaffolding and support to mediate other protein-protein interactions. Cdc27 and Cdc23 have been shown to support the binding of Cdc20 and Cdh1, as mutations in key residues of these subunits led to increased dissociation of the activators. Apc10/Doc1, has been shown to promote substrate binding by mediating their interactions with Cdh1 and Cdc20.[11] In particular, CDC20 (also known as p55CDC, Fizzy, or Slp1) inactivates CDK1 via ubiquitination of B-type cyclins. This results in activation of Cdh1(a.k.a. Fizzy-related, Hct1, Ste9, or Srw1), which interacts with APC during late mitosis and G1/G0. Cdh1 is inactivated via phosphorylation during S, G2 and early M phase. During these points in the cycle, it is not able to be assembled.[12] Evidence shows that APC3 and APC7 serve to recruit Cdh1 to the anaphase-promoting complex.[13] This further supports that Cdh1 is responsible for maintaining APC activity during G1. Cdh1 does not require APC to be phosphorylated in order to bind, in fact, phosphorylation of Cdh1 by Cdks prevents it from binding to APC from S to M phase. With destruction of M-Cdk, release of CDC20 from the APC and binding of Cdh1 can now occur, allowing APC activity to continue on during G1 entry.[1] While Cdh1 recognizes M and S cyclins, allowing for their destruction until the entire cell commits to proceed to a new cycle, it does not recognize G1/S cyclins, and during G1/S phase, their cyclin activity can rise unhindered and phosphorylate and thus inactivating Cdh1 and therefore APC. The subunit Apc15 plays an important role in APC/CCdc20 activation following the bi-orientation of sister chromatids across the metaphase plate. When kinetochores are unattached to spindles, mitotic checkpoint complexes (MCC) and inhibit APC. In the absence of Apc15, MCCs and Cdc20 remain locked on the APC/C preventing its activity once the spindle checkpoint requirements are met. Apc15 mediates the turnover of Cdc20 and MCCs to provide information on the attachment state of kinetochores.[14] CDC27/APC3One of the subunits that exhibit the TPR motif, CDC27 has been identified to interact with mitotic checkpoint proteins such as Mad2, p55CDC and BUBR1, suggesting that it may have involvement in the timing of M phase.[15] Evidence shows that CDC27 is involved in a ternary complex with SMAD2/3 and Cdh1, which is created in response to TGFβ signalling. Because of its interaction with Cdh1 in particular, it has a potential role in determining affinity between APC and its activators Cdc20 and Cdh1. A study suggests that TGF-β-induced Cdc27 phosphorylation enhances interaction between cdc27 and Cdh1–which is directly involved in activating APC.[16] CDC27 can serve as a vehicle through which TGFβ signalling can activate APC. Induced CDC27 hyperphosphorylation by TGFβ showed elevated APC activity. CDC23, CDC16, CDC27CDC23, another TPR subunit interacts with SWM1, binding to the D-box of CLB2. Based upon hybrid assays in vivo and co-immunoprecipitation in vitro, it is suggested that Cdc16p, Cdc23p and Cdc27p (analogs in Sacchyromyces cerevisiae) interact and form a macromolecular complex. Their common theme of TPR is suggested to mediate these interactions.[17] As for Cdc27 and Cdc16 in drosophila, their functions have been tested via RNA interference (RNAi).[18] Results suggest that they may mediate activity of the entire complex via different mechanisms at different sites. In further drosophila studies, Cdk16 and cdk23 appear to be activated via phosphorylation by Polo-like kinase 1 (Plk1) and its fission yeast counterpart, appear to bind particularly to Cdc23.[19] The complex is understood to be regulated by activators CDC20 and Cdh1 during mitosis. Their role in degradation for cyclin B is demonstrated by a screen of Saccharomyces cerevisiae mutants defective for cyclin B degradation, which were found to have mutations in CDC16 and CDC23 genes. Mutants for CDC27, CDC23 and CDC 27 all resulted in a cell-cycle arrest at metaphase.[20] Substrate recognitionAPC/C substrates have recognition amino acid sequences that enable the APC/C to identify them. The most common sequence is known as the destruction box or D-box. APC/C brings together an E2 ubiquitin-conjugating enzyme and the D-box rather than being an intermediate covalent carrier.[21] The D-box should have a version of the following amino acid sequence: RXXLXXXXN, where R is arginine, X is any amino acid, L is Leucine, and N is asparagine. The Ken-box is another motif of importance. Its sequence should resemble the one that follows: KENXXXN, where K is lysine and E is glutamate. The last amino acid position in the Ken-box is highly variable. Though it has been shown that mutations in the sequences do inhibit destruction of the proteins "in vivo", there is still much to learn about how proteins are targeted by the APC/C.[1] Once bound to APC/C, Cdc20 and Cdh1 serve as D and KEN box receptors for various APC substrates. Kraft et al. have shown that the substrates' D boxes bind directly to the highly conserved WD40 repeat propeller region on the APC activators. It is important to note that the conserved area of the propeller of Cdh1 is much larger than that of Cdc20, allowing Cdh1 to have a broader substrate specificity, consistent with the fact that APC/CCdh1 also activates APC-mediated destruction of KEN box containing substrates. The D box further enhances protein degradation, for Lysine residues in close proximity to the D box serve as targets of ubiquitylation. It has been found that a Lys residue immediately C-terminal to the D box can function as a ubiquitin acceptor.[22] Many APC substrates contain both D and KEN boxes, with their ubiquitylation by either APC/CCdc20 or APC/CCdh1 dependent on both sequences, yet some substrates contain only either a D box or a KEN box, in one or multiple copies. Having two distinct degradation sequences creates a high level of substrate specificity on the APC/C, with APC/CCdc20 being more dependent on the D box and APC/CCdh1 more dependent on the KEN box. For example, APC/CCdh1 is capable of ubiquitylating KEN box-only-containing substrates like Tome-1 and Sororin.[6] Although Cdc20 and Cdh1 may serve as D and KEN box receptors, the low affinity of these co-activator–substrate interactions suggests that it is unlikely that the co-activators alone are sufficient to confer high-affinity substrate binding to the APC/CCdc20 and APC/CCdh1.[6] Consequently, core APC/C subunits, like Apc10, contribute towards substrate association as well. In APC/C constructs lacking the Apc10/Doc1 subunit, substrates like Clb2 are unable to associate with APCΔdoc1–Cdh1, while addition of purified Doc1 to the APCΔdoc1–Cdh1 construct restores the substrate binding ability.[11] Metaphase to anaphase transitionAs metaphase begins, the spindle checkpoint inhibits the APC/C until all sister-kinetochores are attached to opposite poles of the mitotic spindle, a process known as chromosome biorientation. When all kinetochores are properly attached, the spindle checkpoint is silenced and the APC/C can become active. M-Cdks phosphorylate subunits on the APC/C that promote binding to Cdc20. Securin and M cyclins (cyclin A and cyclin B) are then targeted by APC/CCdc20 for degradation. Once degraded, separin is released, cohesin is degraded and sister chromatids are prepared to move to their respective poles for anaphase.[1] It is likely that, in animal cells, at least some of the activation of APC/CCdc20 occurs early in the cell cycle (prophase or prometaphase) based on the timing of the degradation of its substrates. Cyclin A is degraded early in mitosis, supporting the theory, but cyclin B and securin are not degraded until metaphase. The molecular basis of the delay is unknown, but is believed to involve the key to the correct timing of anaphase initiation. In animal cells the spindle checkpoint system contributes to the delay if it needs to correct the bi-orientation of chromosomes. Though how the spindle checkpoint system inhibits cyclin B and securin destruction while allowing cyclin A to be degraded is unknown. The delay may also be explained by unknown interactions with regulators, localization and phosphorylation changes.[1] This initiates a negative feedback loop. While activation of APC/CCdc20 requires M-Cdk, the complex is also responsible for breaking the cyclin to deactivate M-CdK. This means that APC/CCdc20 fosters its own deactivation. It is possible that this negative feedback is the backbone of Cdk activity controlled by M and S cyclin concentration oscillations.[1] M to G1 transitionUpon completion of mitosis, it is important that cells (except for embryonic ones) go through a growth period, known as G1 phase, to grow and produce factors necessary for the next cell cycle. Entry into another round of mitosis is prevented by inhibiting Cdk activity. While different processes are responsible for this inhibition, an important one is activation of the APC/C by Cdh1. This continued activation prevents the accumulation of cyclin that would trigger another round of mitosis and instead drives exit from mitosis.[1] In the beginning of the cell cycle Cdh1 is phosphorylated by M-Cdk, preventing it from attaching to APC/C. APC/C is then free to attach to Cdc20 and usher the transition from metaphase to anaphase. As M-Cdk gets degraded later in mitosis, Cdc20 gets released and Cdh1 can bind to APC/C, keeping it activated through the M/G1 transition. A key difference to note is that while binding of Cdc20 to APC/C is dependent on phosphorylation of APC/C by mitotic Cdks, binding of Cdh1 is not. Thus, as APCCdc20 becomes inactivated during metaphase due to dephosphorylation resulting from inactive mitotic Cdks, Cdh1 is able to immediately bind to APC/C, taking Cdc20's place. Cdc20 is also a target of APC/CCdh1, ensuring that APC/CCdc20 is shut down. APC/CCdh1 then continues working in G1 to tag S and M cyclins for destruction. However, G1/S cyclins are not substrates of APC/CCdh1 and therefore accumulate throughout this phase and phosphorylate Cdh1. By late G1, enough of the G1/S cyclins have accumulated and phosphorylated Cdh1 to inactivate the APC/C until the next metaphase.[1] Once in G1, APCCdh1 is responsible for the degradation of various proteins that promote proper cell cycle progression. Geminin is a protein that binds to Cdt1 which prevents its binding to the origin recognition complex (ORC). APCCdh1 targets geminin for ubiquitination throughout G1, keeping its levels low. This allows Cdt1 to carry out its function during pre-RC assembly. When APCCdh1 becomes inactive due to phosphorylation of Cdh1 by G1/S cyclins, geminin activity is increased again. Additionally, Dbf4 stimulates Cell division cycle 7-related protein kinase (Cdc7) activity, which promotes activation of replication origins. APCCdh1 is thought to target Dbf4 for destruction. This could provide an answer as to how Cdc7 is activated at the beginning of a new cell cycle. Its activity likely corresponds to the inactivation of APC/CCdh1 by G/S cyclins.[1] Additional regulationAPC/CCdc20 inactivation during early stages of the cell cycle is partially achieved by the protein Emi1. Initial experiments have shown that addition of Emi1 to Xenopus cycling extracts can prevent the destruction of endogenous cyclin A, cyclin B, and mitotic exit, suggesting that Emi1 is able to counteract the activity of the APC. Furthermore, depletion of Emi1 in somatic cells leads to the lack of accumulation of cyclin B. The lack of Emi1 likely leads to a lack of inhibition of the APC preventing cyclin B from accumulating.[23] From these early observations, it has been confirmed that in G2 and early mitosis, Emi1 binds and inhibits Cdc20 by preventing its association with APC substrates. Cdc20 can still be phosphorylated and bind to APC/C, but bound Emi1 blocks Cdc20's interaction with APC targets.[1] Emi1 association with Cdc20 allows for the stabilization of various cyclins throughout S and G2 phase, but Emi1's removal is essential for progression through mitosis. Thus, in late prophase, Emi1 is phosphorylated by Polo-like kinase, Plk. Plk is activated during early mitosis by Cdk1 activity, and its phosphorylation of Emi1's BTRC (gene) βTrCP binding site makes it a target for SCF, leading to its subsequent destruction in prometaphase.[24] Emi1's destruction leads APC/CCdc20 activation, allowing for the destruction of cyclin A in early mitosis. Emi1 levels begin to rise again in G, which help inhibit APC/CCdh1.[1] Regulation of APC/CCdc20 activity towards metaphase substrates like securin and cyclin B may be a result of intracellular localization. It is hypothesized that spindle checkpoint proteins that inhibit APC/CCdc20 only associate with a subset of the Cdc20 population localized near the mitotic spindle. In this manner, cyclin A can be degraded while cyclin B and securin are degraded only once sister chromatids have achieved bi-orientation.[1] See alsoReferences
Further reading
External links
|